Current research
Quantum computing
Development of quantum algorithms for quantum chemistry and quantum many-body physics, including ground state, excited state, nonadiabatic (both mixed quantum-classical and quantum) dynamics, open quantum systems, and beyond
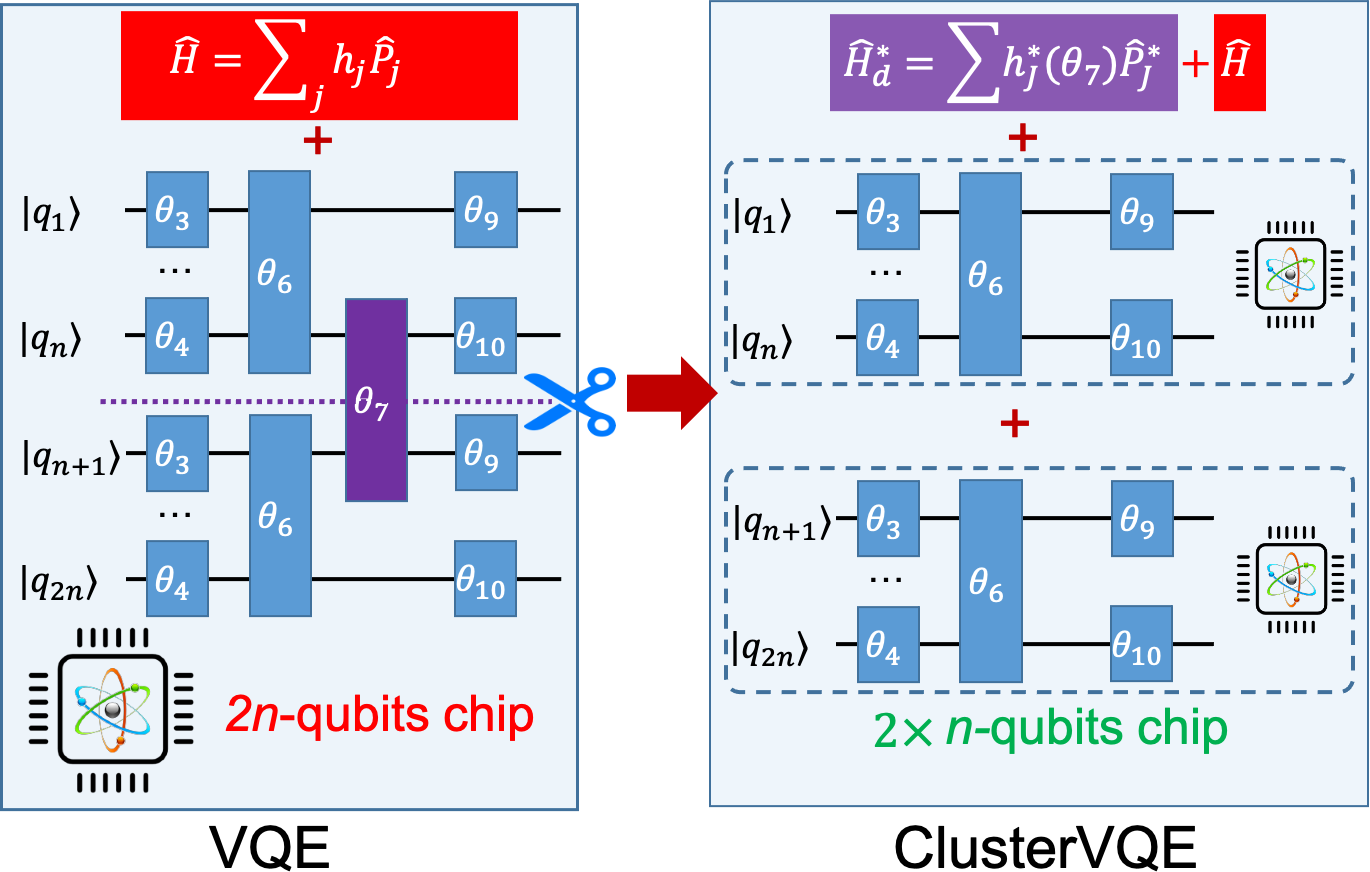
Quantum computers were long envisioned as game changers for chemistry and materials modeling, but truly efficient quantum algorithms remain elusive. Our team is developing circuit-efficient, noise-resilient quantum algorithms to (i) estimate energies in quantum many-body systems and (ii) simulate quantum dynamics in complex environments on forthcoming fault-tolerant quantum computers, while identifying where genuine quantum advantage may emerge in quantum many-body physics.
References:
- J. Chem. Phys. 164, 084103 (2026)
- J. Chem. Theory Comput. 21, 11335 (2025)
- arXiv:2512.14842
- arXiv:2511.13038
- arXiv:2510.21075
- arXiv:2406.08675
- arXiv:2406.06625
- J. Chem. Theory Comput. 19, 9136 (2024)
- Chem. Sci. 14, 2405 (2023)
- Sci. Rep. 12, 16824 (2022)
- J. Chem. Theory Comput. 18, 9, 5312 (2022)
- npj Quantum Info. 8, 96 (2022)
- J. Chem. Theory Comput. 18, 4177 (2022)
- Sci. Rep. 11, 18796 (2021)
- PRX Quantum 2, 020337 (2021)
Cavity quantum chemistry
Develop multiscale theory and corresponding massively parallel algorithms for molecular quantum electrodynamics
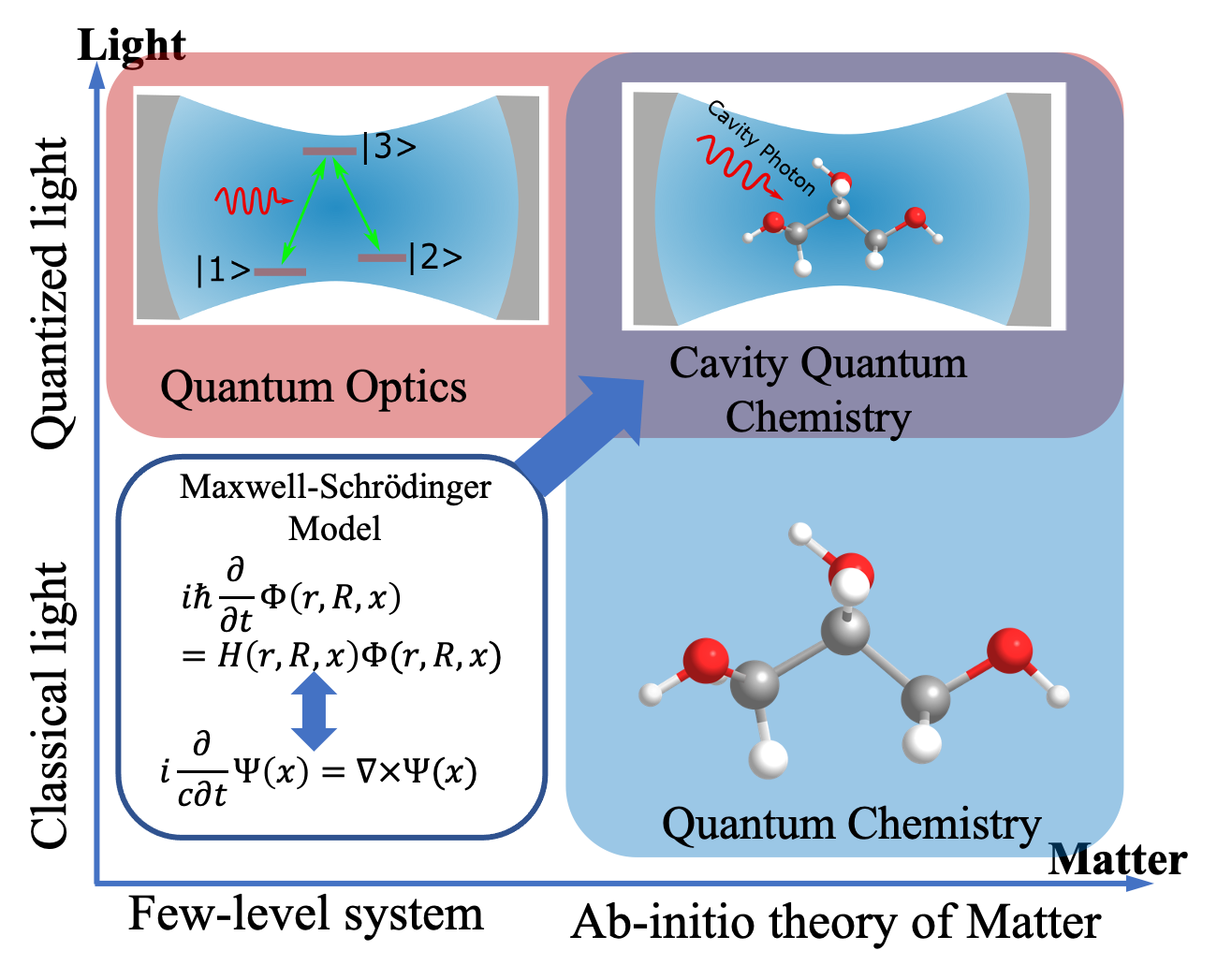
The polaritons formed due to the strong light-matter interaction are able to alter the molecular properties (polaritonic chemistry), which provides a promising way to establish coupled spin-polariton for achieving quantum transduction. However, the underlying mechanisms are unclear and require state-of-the-art ab initio modeling of polaritonic chemistry. Such modeling remains a big challenge because it involves multiple degrees of freedom and interactions across different time and length scales. While current theories (quantum optics and quantum chemistry) are good at treating the light-matter interaction in separate domains by simplifying either light or matter, there is no reliable and computationally feasible ab initio framework that can be used to predict emergent properties of polaritonic chemistry. To this aim, we aim to deliver new theoretical frameworks and ab initio quantum many-body modeling capabilities for polaritonic chemistry, which treats electronic and photonic degrees of freedom on an equal footing. Efficient numerical algorithms will be developed by leveraging our ongoing experience in exascale computing development. Modular programming will be adopted to enhance code’s maintainability, portability, and reusability on emergent computing architectures.
References:
- J. Chem. Theory Comput. 22, 1770 (2026)
- J. Chem. Theory Comput. 21, 10035 (2025)
- arXiv:2411.15022
- arXiv:2410.16478
- Phys. Rev. A. 109, 032804 (2024)
- J. Chem. Theory Comput. 20, 891 (2024)
- Nat. Commun., 8035 (2023)
- arXiv:2310.18228
- Phys. Chem. Chem. Phys. 25, 31554 (2023)
- J. Chem. Phys. 151, 154109 (2019).
Cavity quantum materials
Light-induced quantum states and phase transition in low-dimensional materials: where Quantum materials meet cavities
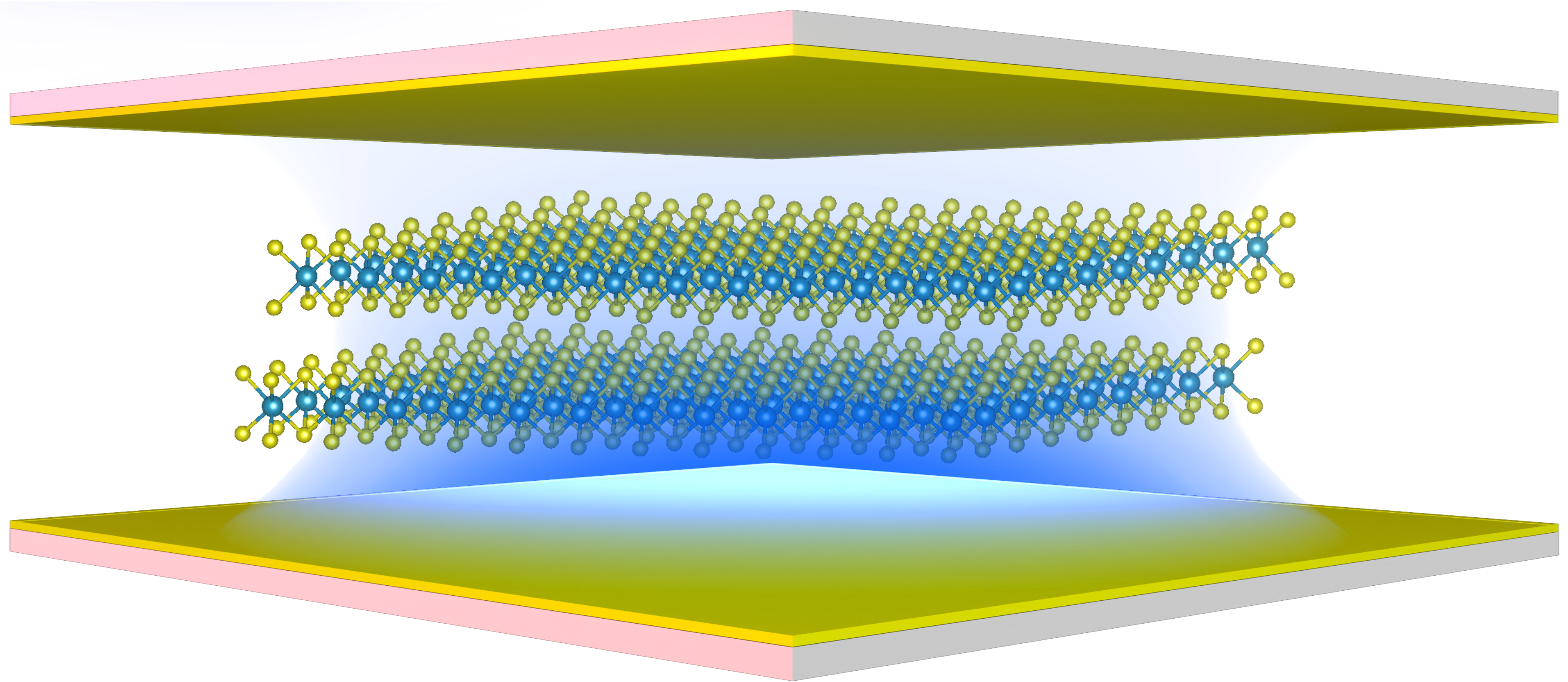
Cavity Quantum Materials lie at the intersection of quantum materials and cavity QED, where the interplay between light and matter gives rise to emergent many-body phenomena. Our research focuses on developing and applying quantum many-body and multi-scale methods to uncover novel quantum phases, collective excitations, and non-equilibrium effects in materials embedded in cavities. By exploring how strong light-matter interactions modify electronic, magnetic, and topological properties, we aim to pave the way for engineered quantum states and functionalities in correlated materials.
References:
- Phys. Rev. A 113 (3), 033730
- arXiv:2601.03230
- Phys. Rev. A (2026)
- arXiv:2411.15022
Tensor network and nuclear quantum dynamics
Development of efficient tensor network methods for quantum dynamics
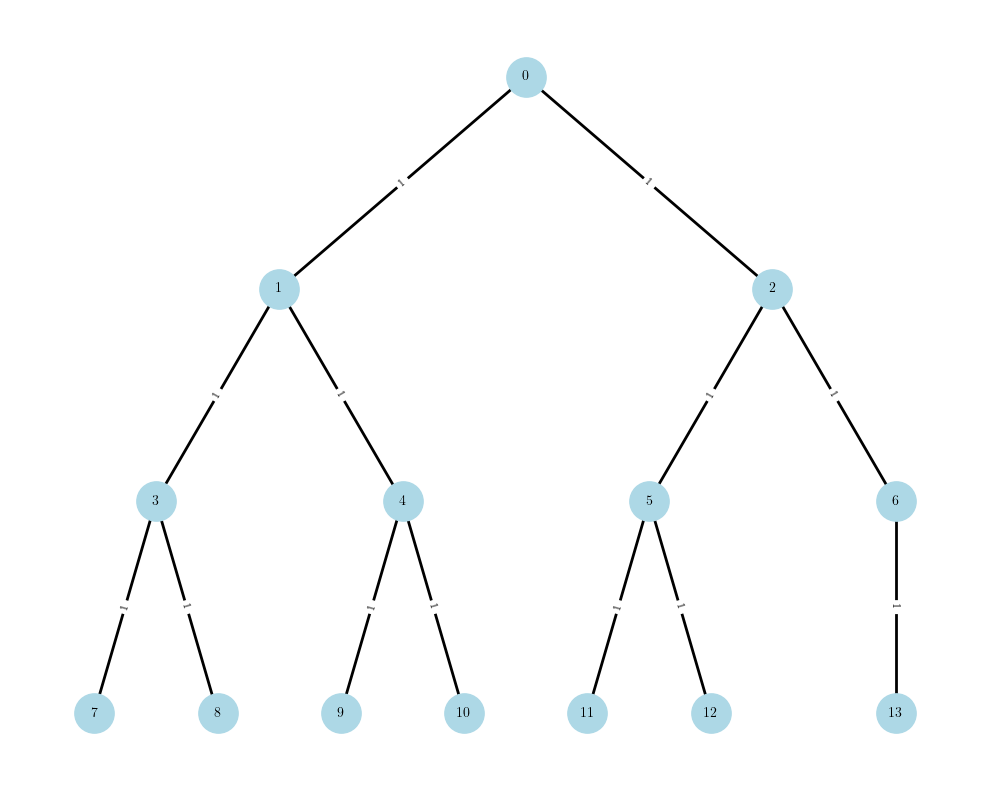
We aim to develop efficient tree tensor network (TTN)-based quantum dynamics methods for both closed and open quantum system.
References:
- TBA.
Plasmon physics & chemistry
Develop and apply theoretical models and numerical methods to understand the physics of plasmon-mediated chemistry
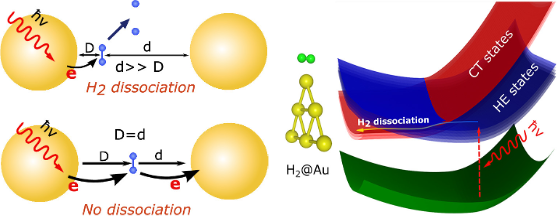
Plasmon chemistry is an emerging field that investigates the interactions between plasmonic excitations and chemical processes, with potential applications in areas such as energy conversion, photocatalysis, sensing, and nanotechnology. In this project, we focus on advancing the understanding of plasmon chemistry by developing a comprehensive physical model, first-principles methods, and non-adiabatic dynamics to elucidate the underlying mechanisms governing plasmon-driven chemical reactions. Our work encompasses the systematic investigation of localized surface plasmon resonances (LSPRs) and their influence on chemical processes at the nanoscale. By employing state-of-the-art computational techniques and rigorous theoretical frameworks, we aim to provide a fundamental understanding of the interplay between plasmonic excitations, electron transfer, and reactive pathways. This knowledge will serve as a foundation for the rational design of advanced plasmonic materials and devices, with potential applications in diverse fields such as photocatalysis, sensing, and energy conversion.
References:
- Chem. Sci. 14, 4714 (2023)
- J. Chem. Phys. 157, 214201 (2022)
- J. Chem. Phys. 157, 084114 (2022)
- J. Phys. Chem. A 125, 9201 (2021)
- J. Am. Chem. Soc. 142, 13090 (2020)
- Acs Nano 12, 8415 (2018)
- J. Phys. Chem. Lett. 7, 1852 (2016).
Molecular quantum information science
Spin dynamics in the presence of spin-spin and spin-phonon interactions and quantum information transduction
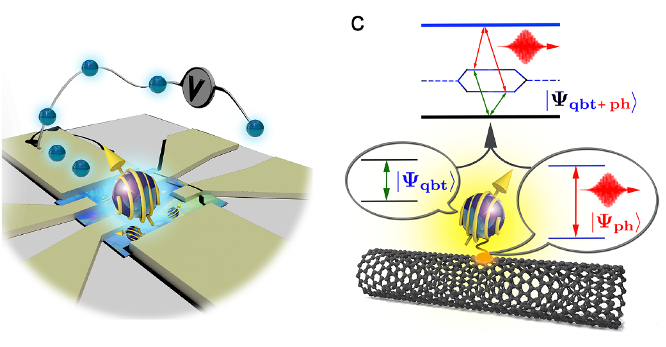
Due to their unique properties and advantages over other qubit systems, molecular qubits have emerged as promising candidates for quantum information processing and quantum computing technologies. The synthetic versatility of molecular compounds allows for the tailoring of magnetic properties, enabling the design of materials with specific characteristics for various applications. Moreover, molecular qubits often exhibit long coherence times and can be integrated into scalable architectures, making them suitable for developing advanced quantum devices. However, understanding the relaxation dynamics of molecular qubits, particularly the spin-spin and spin-phonon interactions, is a crucial challenge that must be addressed to harness their potential fully. Traditional first-principles methods for simulating these interactions can be computationally expensive.
In this research project, we focus on developing a first-principles-based method that employs a quantum embedding technique to tackle these challenges. This approach significantly reduces the computational cost while maintaining high accuracy, allowing for a deeper understanding of the relaxation dynamics of molecular qubits. By efficiently modeling the complex interplay between spin-spin and spin-phonon interactions, our method will not only pave the way for substantial advancements in quantum information processing but also enable the exploration of previously intractable problems in studying molecular qubits. The development of such a method is essential for realizing the full potential of molecular qubits and fostering the growth of quantum information technologies.
References:
- J. Chem.Phys. 163, 224110 (2025)
- arXiv preprint arXiv:2407.08043
- arXiv preprint arXiv:2407.07843.
Previous research
DOE Exascale computing project

In our work, we focus on developing scalable and universal libraries for electronic structure, quantum molecular dynamics, and light-matter interactions, harnessing the power of exascale computing to push the boundaries of what is possible in these fields. By optimizing our libraries for massively parallel and heterogeneous architectures, we aim to provide the research community with efficient and versatile tools for conducting large-scale simulations and investigating complex problems in materials science and quantum physics. This research has the potential to accelerate scientific discovery and serves as a critical step in realizing the full potential of exascale computing for advancing knowledge and innovation.
References:
- CiSE 26, 43 (2024)
- J. Chem. Phys. 160, 122501 (2024).
Bioinspired artificial ion pump
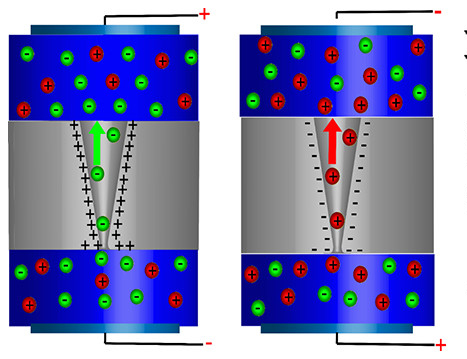
Biological ion channels, the nanoscale gatekeepers in cells, have inspired the creation of artificial counterparts, owing to their essential role in cellular communication and interaction with external environments. Leveraging their potential could spur advancements in numerous fields, including nanotechnology, biomedical devices, and environmental studies. In my research, I developed theoretical models and multiphysics methods to simulate ion transport and pump mechanisms in various artificial channels, encompassing light-driven Ca2+ pump, protein pump and conical pores, showcasing its application in seawater desalination. Additionally, I utilized the developed tools to explore the underlying molecular mechanisms further, laying the groundwork for the emergence of more efficient and innovative artificial ion pumps.
References:
- J. Phys. Chem. B, 119, 15110–15117 (2015)
- J. Phys. Chem. C, 120, 14495–14501 (2016)
- J. Phys. Chem. Lett., 8, 2842 (2017)
- J. Phys. Chem. C, 121, 161 (2017).
Transient and dissipative quantum transport
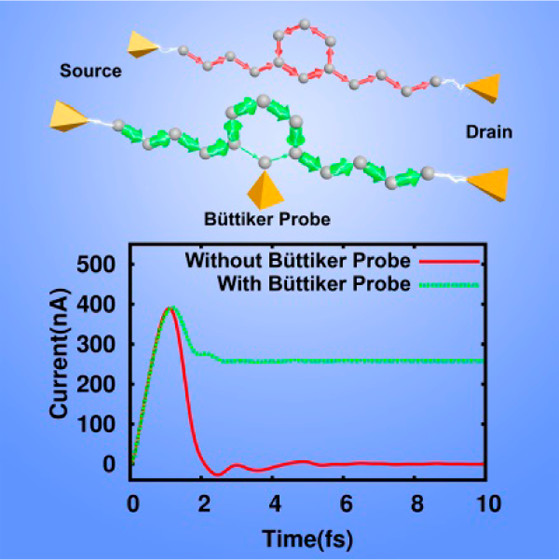
I developed the time-dependent non-equilibrium Green’s function (NEGF) methods to study for studying light-driven quantum transport phenonemna. The method has been used to understand the ultrafast control of currents through a SiO$_2$-based nanoscale, which recovered the main experimental observations and offers a simple and intuitive picture of the effect of ultrafast light control of currents along nanojunctions. This method was used by my collaborators to successfully predict the real and virtual charges in quantum transport, which was confirmed in a recent experiment [Nature, 605, 251–255 (2022)].
References:
- Nanoscale, 5, 169 (2013)
- Phys. Rev. B, 87, 085110 (2013)
- J. Chem. Phys., 138, 164121 (2013)
- Phys. Status Solidi B 250, 2481 (2013)
- J. Phys. Chem. Lett., 5, 2748 (2014)
- J. Chem. Phys., 142, 164101 (2015)
- J. Chem. Phys. 143, 104112 (2015)
- arXiv:1510.02960
- Nat. Commun., 9, 2070 (2018).
Quantum-mechanical (QM) and Multiscale QM/EM simulation of (opto)electronics

Within the framework of the Non-equilibrium Green’s Function (NEGF) method and many-body perturbation theory, I developed efficient quantum mechanical approaches for simulating nano photovoltaics and light-emitting devices. By developing fast and highly efficient massively parallel algorithms, my method allows the simulation of optoelectronic properties of realistic devices with dozens of thousands of atoms on a few computational nodes. The method has also been extended to study the thin-film devices and extended to integrate with the Maximally Localized Wannier Function-based Tight-Binding (TB) method. Besides, I further developed a multiscale quantum mechanics/electromagnetics method to model the optoelectronics in complex electromagnetic (EM) environments, such as plasmon-enhanced solar cells.
References:
- J. Phys. Chem. Lett., 5, 1272 (2014)
- Chem. Soc. Rev., 44, 1763 (2015)
- J. Phys. Chem. Lett., 6, 4410 (2015)
- PIER 154, 163 (2015)
- J. Comput. Phys. 286, 49 (2015)
- Nanoscale, 8, 13168–13173 (2016)
- J. Phys. Chem. Lett., 8, 571 (2017)
- J. Phys. Chem. Lett., 9, 6915 (2018)
- J. Phys. Chem. C, 123, 15761 (2019)